













3'30" 읽기
- Telomere-to-Telomere(T2T) 컨소시엄이 우리 게놈의 나머지 격차를 줄이는 데 성공
- 인간 게놈 최초 해독된 지 21년 만에 전체 인간 DNA 코드가 처음으로 완전히 해독돼
- "새로운 참조 게놈은 단일 염기 수준까지 매우 정확해 수십만 개의 변이체를 감지할 수 있다"
2001년 인간 게놈 프로젝트는 최초로 인간 게놈 해독에 성공해 의학 및 유전학의 이정표가 되었다. 그러나 이 첫 번째 참조 게놈은 완전하지 않았다. DNA 서열의 약 8%를 시퀀싱할 수 없었다. 여기에는 주로 염색체 끝에 있는 유전 물질의 부분과 염색체의 중심 마디인 중심절(centromere)이 포함된다.
이유:
이 섹션의 DNA 시퀀스는 거의 동일한 사본과 반복으로 구성됐다.
현재의 시퀀싱 방법은 게놈을 수백 염기 길이의 짧은 단편으로 분해한 다음 나중에 올바르게 재조립해야 한다. 그러나 이 조각들이 거의 동일하다면 그것은 불가능하다. 같은 색깔의 조각들로 이루어진 퍼즐과 비슷하다.
생명책의 새로운 장
이제 바뀌었다. 시퀀싱 기술의 발전 덕분에 Telomere-to-Telomere(T2T) 컨소시엄이 이제 우리 게놈의 나머지 격차를 줄이는 데 성공했다. 염색체 끝에서 중심체까지 인간 게놈의 전체 DNA 서열을 완전히 분해하여 보여주는 참조 게놈이 처음으로 사용 가능하다.
워싱턴 대학의 에반 아이슬러(Evan Eichler)는 "지금 우리는 이전에 결코 읽을 수 없었던 생명책의 장(chapter)들을 보고 있다"고 말했다. T2T-CHM13으로 명명된 참조 게놈은 이전에 읽을 수 없었던 약 2억 개의 염기 서열을 보여줌으로써 우리 게놈의 이전에 "어두웠던" 8%를 처음 엿볼 수 있게 해준다. 또한 새로운 디코딩은 이전 참조 게놈의 수천 가지 구조적 오류를 수정한다.
두 가지 방법을 결합
이 돌파구는 두 가지 새로운 시퀀싱 시스템의 조합으로 가능했다.
소위 Oxford nanopore sequencing은 중간 정도의 정확도에도 불구하고 최대 100만 염기 길이의 DNA 섹션을 읽을 수 있다. 개별 DNA 분자는 좁은 구멍을 통해 통과하고 장치는 DNA 염기에 따라 발생하는 미묘한 전압 변화를 등록한다. 이 시스템은 2020년 처음으로 여성 X 염색체를 완전히 매핑하는 데 사용되었다.
T2T 컨소시엄의 과학자들은 이 방법을 두 번째 방법과 결합했다.
Pacific Biosciences의 시스템은 길이가 약 2만 개 염기인 세그먼트를 생성하지만 99% 정밀도로 읽을 수 있다. 이것은 인간 게놈의 모든 누락된 부분을 완전하고 높은 정밀도로 해독하는 것을 가능하게 했다. 시퀀싱에 사용된 유전 물질은 우연히 모든 자매 염색체가 한 부모에게서만 나온 인간 세포주에서 유래했다. 이것은 시퀀싱을 더 쉽게 만들었다고 팀은 설명했다.
새로운 유전자 및 유전자 변이체
새로운 시퀀싱은 이제 삶의 청사진과 우리를 인간으로 만드는 코드에 대한 완전히 새로운 통찰력을 열어준다. 연구자들은 새로 해독된 DNA 세그먼트 중에서 이전에 알려지지 않은 99개의 단백질 코딩 유전자와 거의 2천 개의 다른 유전자 후보를 이미 확인했다. 전에 알려지지 않은 많은 유전자 변이체도 현재 나타나고 있다.
"새로운 참조 게놈은 단일 염기 수준까지 매우 정확하여 수십만 개의 변이체를 감지할 수 있다"고 산타크루즈에 있는 캘리포니아 대학의 Karen Miga가 설명한다. "이러한 새로운 변이체 중 많은 부분이 질병에 기여하는 것으로 알려진 유전자에 있다." 이를 아는 것은 많은 질병의 유전적 기초를 더 잘 이해할 수 있는 새로운 기회를 열어준다.
중심체에 대한 첫 번째 통찰력
또한 염색체의 두 반쪽을 함께 유지하는 접합부인 중심체의 구조에 대한 새로운 통찰력이 중요하다. 그들은 이 자매 염색분체를 분리하는 감수 분열인 감수 분열에서 중요한 역할을 한다. "감수 분열의 이 단계가 잘못되면 유산이나 유전 질환을 일으키는 염색체 이상이 발생할 수 있다"고 버클리 캘리포니아 대학의 니콜라스 알테모세(Nicolas Altemose)가 설명했다. 암은 또한 이러한 조절 장애의 결과일 수 있다.
이러한 기형의 원인을 식별할 수 있으려면 중심체와 그 변이체의 유전 코드를 아는 것이 더욱 중요하다. 이것이 바로 새로운 T2T 참조 게놈 덕분에 가능한 일이다. “이전에는 그곳에 무엇이 숨겨져 있는지에 대한 매우 흐릿한 그림만 가지고 있었다. 그러나 이제는 개별 DNA 염기까지 명확해졌다”고 Altemose는 말했다.
연구는 계속
최초의 인간 게놈 프로젝트의 공동 작업자 중 한 명인 워싱턴 대학의 밥 워터스턴은 "인간 게놈의 최초의 완전한 해독은 중요한 이정표"라고 말했다. "우리는 20년 전에 이것을 하고 싶었지만 그 당시에는 기술이 준비되지 않았다.“
T2T 컨소시엄의 작업은 여기서 끝나지 않는다. 그들은 이미 양쪽 부모에서 유래한 정상적인 염색체 세트로 게놈을 해독하는 작업을 하고 있다. 또한 유사한 정확도와 완전성으로 다른 인구의 사람들의 게놈을 시퀀싱하기를 원한다. 이것은 다른 인간 유형의 유사점, 차이점 및 진화에 대한 새로운 통찰력을 제공할 수 있다.
(Science, 2022; doi:10.1126/science.abp8653; doi:10.1126/science.abj6987 및 기타)
출처: Johns Hopkins University, NIH/National Human Genome Research Institute, Howard Hughes Medical Institute, University of California Santa Cruz, University of California Berkeley
- Telomere-to-Telomere(T2T) 컨소시엄이 우리 게놈의 나머지 격차를 줄이는 데 성공
- 인간 게놈 최초 해독된 지 21년 만에 전체 인간 DNA 코드가 처음으로 완전히 해독돼
- "새로운 참조 게놈은 단일 염기 수준까지 매우 정확해 수십만 개의 변이체를 감지할 수 있다"
최초의 완전한 인간 유전자 코드
최초의 인간 게놈 DNA 염기 서열 완전 해독
유전학의 이정표:
인간 게놈이 최초로 해독된 지 21년 만에 전체 인간 DNA 코드가 이제 처음으로 완전히 시퀀싱되었다. 새로운 참조 게놈은 이전에 너무 많은 코드 반복으로 인해 시퀀싱할 수 없었던 게놈의 8%를 보완한다. 이것은 과학자들이 "Science"의 6개 전문 기사에서 보고한 바와 같이 유전자 조절, 유전적 변이의 범위 및 질병의 원인에 대한 새로운 통찰력을 제공한다.
 |
| ▲ 처음으로 인간 게놈의 완전하고 손상되지 않은 DNA 코드가 알려져 있다. © ktsimage/ iStock |
2001년 인간 게놈 프로젝트는 최초로 인간 게놈 해독에 성공해 의학 및 유전학의 이정표가 되었다. 그러나 이 첫 번째 참조 게놈은 완전하지 않았다. DNA 서열의 약 8%를 시퀀싱할 수 없었다. 여기에는 주로 염색체 끝에 있는 유전 물질의 부분과 염색체의 중심 마디인 중심절(centromere)이 포함된다.
이유:
이 섹션의 DNA 시퀀스는 거의 동일한 사본과 반복으로 구성됐다.
현재의 시퀀싱 방법은 게놈을 수백 염기 길이의 짧은 단편으로 분해한 다음 나중에 올바르게 재조립해야 한다. 그러나 이 조각들이 거의 동일하다면 그것은 불가능하다. 같은 색깔의 조각들로 이루어진 퍼즐과 비슷하다.
생명책의 새로운 장
이제 바뀌었다. 시퀀싱 기술의 발전 덕분에 Telomere-to-Telomere(T2T) 컨소시엄이 이제 우리 게놈의 나머지 격차를 줄이는 데 성공했다. 염색체 끝에서 중심체까지 인간 게놈의 전체 DNA 서열을 완전히 분해하여 보여주는 참조 게놈이 처음으로 사용 가능하다.
워싱턴 대학의 에반 아이슬러(Evan Eichler)는 "지금 우리는 이전에 결코 읽을 수 없었던 생명책의 장(chapter)들을 보고 있다"고 말했다. T2T-CHM13으로 명명된 참조 게놈은 이전에 읽을 수 없었던 약 2억 개의 염기 서열을 보여줌으로써 우리 게놈의 이전에 "어두웠던" 8%를 처음 엿볼 수 있게 해준다. 또한 새로운 디코딩은 이전 참조 게놈의 수천 가지 구조적 오류를 수정한다.
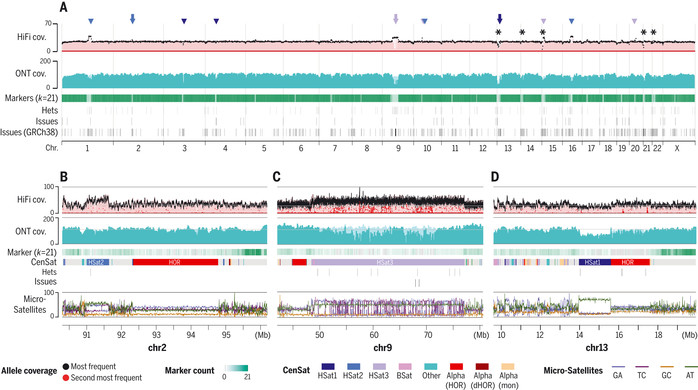 |
| ▲ Fig. 3. 시퀀싱 적용 범위 및 어셈블리 유효성 검사.(A) 매핑된 HiFi 및 ONT 읽기의 균일한 전체 게놈 적용 범위는 밝은 음영의 기본 정렬과 어두운 음영의 오버레이된 마커 지원 정렬로 표시된다. 큰 HSat 어레이(30)는 화살표로 표시된 삽입 영역과 별표로 표시된 rDNA 어레이의 위치가 있는 삼각형으로 표시. 고유한 마커 빈도가 낮은 영역(연한 녹색)은 고유한 마커 밀도의 감소에 해당하지만 신뢰도가 낮은 기본 정렬에 의해 복구된다. 주석이 달린 어셈블리 문제는 T2T-CHM13 및 GRCh38에 대해 비교된다. Hets, 이형접합 변이체; k, 마커 크기. (B에서 D) 각각 그림 2, B에서 D에 나타난 게놈 영역에 해당하는 확대. 특정 위성 내에서 균일한 적용 범위 변경은 재현 가능하며 시퀀싱 편향으로 인해 발생할 수 있다. 식별된 이형접합 변이체 및 어셈블리 문제는 아래에 표시돼 있으며 일반적으로 1차 대립유전자(검은색)의 낮은 적용 범위와 2차 대립유전자(빨간색)의 증가된 적용 범위에 해당한다. 모든 128bp 창에 대한 미세위성 반복의 백분율이 하단에 표시. (출처: 관련논문 The complete sequence of a human genome / SCIENCE · 31 Mar 2022) |
두 가지 방법을 결합
이 돌파구는 두 가지 새로운 시퀀싱 시스템의 조합으로 가능했다.
소위 Oxford nanopore sequencing은 중간 정도의 정확도에도 불구하고 최대 100만 염기 길이의 DNA 섹션을 읽을 수 있다. 개별 DNA 분자는 좁은 구멍을 통해 통과하고 장치는 DNA 염기에 따라 발생하는 미묘한 전압 변화를 등록한다. 이 시스템은 2020년 처음으로 여성 X 염색체를 완전히 매핑하는 데 사용되었다.
T2T 컨소시엄의 과학자들은 이 방법을 두 번째 방법과 결합했다.
Pacific Biosciences의 시스템은 길이가 약 2만 개 염기인 세그먼트를 생성하지만 99% 정밀도로 읽을 수 있다. 이것은 인간 게놈의 모든 누락된 부분을 완전하고 높은 정밀도로 해독하는 것을 가능하게 했다. 시퀀싱에 사용된 유전 물질은 우연히 모든 자매 염색체가 한 부모에게서만 나온 인간 세포주에서 유래했다. 이것은 시퀀싱을 더 쉽게 만들었다고 팀은 설명했다.
새로운 유전자 및 유전자 변이체
새로운 시퀀싱은 이제 삶의 청사진과 우리를 인간으로 만드는 코드에 대한 완전히 새로운 통찰력을 열어준다. 연구자들은 새로 해독된 DNA 세그먼트 중에서 이전에 알려지지 않은 99개의 단백질 코딩 유전자와 거의 2천 개의 다른 유전자 후보를 이미 확인했다. 전에 알려지지 않은 많은 유전자 변이체도 현재 나타나고 있다.
"새로운 참조 게놈은 단일 염기 수준까지 매우 정확하여 수십만 개의 변이체를 감지할 수 있다"고 산타크루즈에 있는 캘리포니아 대학의 Karen Miga가 설명한다. "이러한 새로운 변이체 중 많은 부분이 질병에 기여하는 것으로 알려진 유전자에 있다." 이를 아는 것은 많은 질병의 유전적 기초를 더 잘 이해할 수 있는 새로운 기회를 열어준다.
중심체에 대한 첫 번째 통찰력
또한 염색체의 두 반쪽을 함께 유지하는 접합부인 중심체의 구조에 대한 새로운 통찰력이 중요하다. 그들은 이 자매 염색분체를 분리하는 감수 분열인 감수 분열에서 중요한 역할을 한다. "감수 분열의 이 단계가 잘못되면 유산이나 유전 질환을 일으키는 염색체 이상이 발생할 수 있다"고 버클리 캘리포니아 대학의 니콜라스 알테모세(Nicolas Altemose)가 설명했다. 암은 또한 이러한 조절 장애의 결과일 수 있다.
이러한 기형의 원인을 식별할 수 있으려면 중심체와 그 변이체의 유전 코드를 아는 것이 더욱 중요하다. 이것이 바로 새로운 T2T 참조 게놈 덕분에 가능한 일이다. “이전에는 그곳에 무엇이 숨겨져 있는지에 대한 매우 흐릿한 그림만 가지고 있었다. 그러나 이제는 개별 DNA 염기까지 명확해졌다”고 Altemose는 말했다.
연구는 계속
최초의 인간 게놈 프로젝트의 공동 작업자 중 한 명인 워싱턴 대학의 밥 워터스턴은 "인간 게놈의 최초의 완전한 해독은 중요한 이정표"라고 말했다. "우리는 20년 전에 이것을 하고 싶었지만 그 당시에는 기술이 준비되지 않았다.“
T2T 컨소시엄의 작업은 여기서 끝나지 않는다. 그들은 이미 양쪽 부모에서 유래한 정상적인 염색체 세트로 게놈을 해독하는 작업을 하고 있다. 또한 유사한 정확도와 완전성으로 다른 인구의 사람들의 게놈을 시퀀싱하기를 원한다. 이것은 다른 인간 유형의 유사점, 차이점 및 진화에 대한 새로운 통찰력을 제공할 수 있다.
(Science, 2022; doi:10.1126/science.abp8653; doi:10.1126/science.abj6987 및 기타)
출처: Johns Hopkins University, NIH/National Human Genome Research Institute, Howard Hughes Medical Institute, University of California Santa Cruz, University of California Berkeley
[더사이언스플러스=문광주 기자]
[저작권자ⓒ the SCIENCE plus. 무단전재-재배포 금지]







주요기사
+



많이 본 기사





Basic Science
+

AI & Tech
+
Photos
+






- the SCIENCE plus (04426) 서울시 용산구 이촌로 88길 30 대표전화 : 010-7145-3730 청소년보호관리책임자 : 문광주
- 발행인· 편집인 : 문광주 등록번호 : 경기 아52382 등록/발행일 : 2019-11-07 제보메일 : helloscienceplus@gmail.com
- 본 콘텐츠의 저작권은 the SCIENCE plus 또는 제공처에 있으며 이를 무단 이용하는 경우 저작권법 등에 따라 법적책임을 질 수 있습니다.
- Copyright ⓒ the SCIENCE plus All rights reserved. 0.0284